
SARCLISA CONCENTRATE FOR SOLUTION FOR INFUSION 20MG/ML [SIN16630P]
Active ingredients: SARCLISA CONCENTRATE FOR SOLUTION FOR INFUSION 20MG/ML
Last updated 21 July 2026
Product Info
SARCLISA CONCENTRATE FOR SOLUTION FOR INFUSION 20MG/ML
[SIN16630P]
Product information
Active Ingredient and Strength | ISATUXIMAB - 20 MG/ML |
Dosage Form | INFUSION, SOLUTION CONCENTRATE |
Manufacturer and Country | SANOFI-AVENTIS DEUTSCHLAND GMBH - GERMANY |
Registration Number | SIN16630P |
Licence Holder | SANOFI-AVENTIS SINGAPORE PTE. LTD. |
Forensic Classification | PRESCRIPTION ONLY MEDICINES |
Anatomical Therapeutic Chemical (ATC) code | L01XC38 |
Prescription-only Medicines with Exemptions for Supply without Prescription | NA |
Indication
4.1 Therapeutic Indications
SARCLISA is indicated:
in combination with pomalidomide and dexamethasone, for the treatment of adult patients with relapsed and refractory multiple myeloma who have received at least two prior therapies including lenalidomide and a proteasome inhibitor and have demonstrated disease progression on the last therapy.
in combination with carfilzomib and dexamethasone, for the treatment of adult patients with relapsed or refractory multiple myeloma who have received one to three prior therapies (see section 5.1 – please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information).
in combination with bortezomib, lenalidomide, and dexamethasone, for the treatment of adult patients with newly diagnosed active multiple myeloma who are not eligible for autologous stem cell transplant (ASCT).
Dosing
4.2 Posology and method of administration
SARCLISA should be administered by a healthcare professional, in an environment where resuscitation facilities are available.
Premedication
Prevention of infusion reaction
Premedication should be used prior to SARCLISA infusion with the following medicinal products to reduce the risk and severity of infusion reactions:
Dexamethasone 40 mg oral or intravenous (or 20 mg oral or intravenous for patients ≥75 years of age): when administered in combination with isatuximab and pomalidomide,
Dexamethasone 20 mg (intravenous on the days of isatuximab and/or carfilzomib infusions, and oral on the other days): when administered in combination with isatuximab and carfilzomib.Acetaminophen 650 mg to 1000 mg oral (or equivalent).
Diphenhydramine 25 mg to 50 mg intravenous or oral (or equivalent [e.g., cetirizine, promethazine, dexchlorpheniramine]). The intravenous route is preferred for at least the first 4 infusions.
The above recommended dose of dexamethasone (oral or intravenous) corresponds to the total dose to be administered only once before the infusion, as part of the premedication and the backbone treatment, before isatuximab and pomalidomide and before isatuximab and carfilzomib administration.
The recommended premedication agents should be administered 15–60 minutes prior to starting a SARCLISA infusion. Patients who do not experience an infusion reaction upon their first 4 administrations of SARCLISA may have their need for subsequent premedication reconsidered.
Management of neutropenia
The use of colony-stimulating factors (e.g. G-CSF) should be considered to mitigate the risk of neutropenia. In the event of grade 4 neutropenia, SARCLISA administration should be delayed until neutrophil count improves to at least 1.0 x 109/L (see section 4.4 – please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information).
Prevention of infection
Antibacterial and antiviral prophylaxis (such as herpes zoster prophylaxis) can be considered during treatment (see section 4.4 – please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information).
Posology
The recommended dose of SARCLISA is 10 mg/kg body weight administered as an intravenous infusion in combination with pomalidomide and dexamethasone (Isa-Pd) or in combination with carfilzomib and dexamethasone (Isa-Kd), or in combination with bortezomib, lenalidomide, and dexamethasone (Isa-VRd).
SARCLISA dosing schedules are provided in Tables 1 and 2:
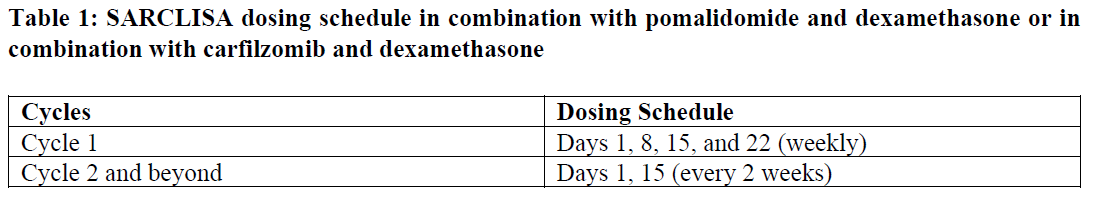
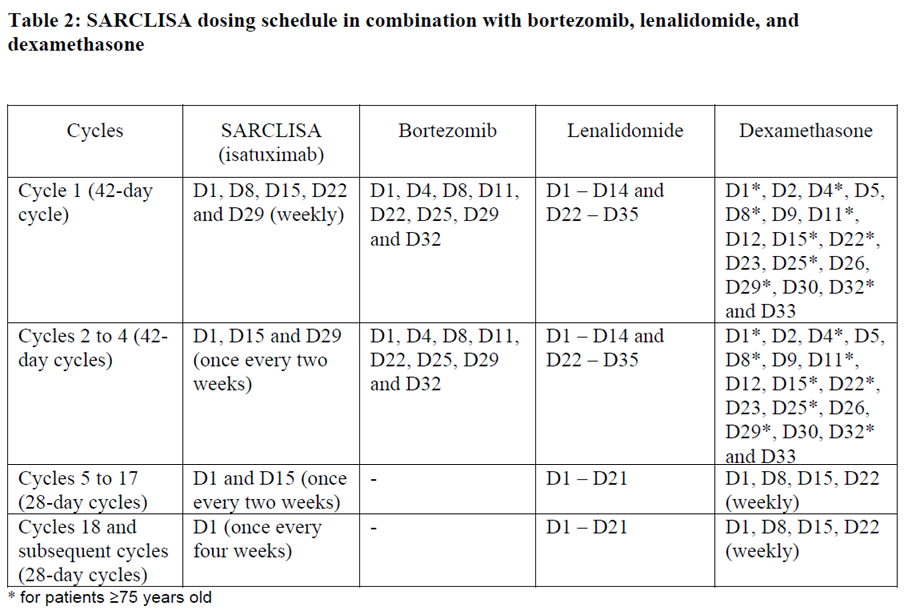
The dose of bortezomib is 1.3 mg/m2. It is administered subcutaneously on days D1, D4, D8, D11, D22, D25, D29 and D32 in the cycles 1 to 4. Bortezomib is not administered from cycle 5.
The dose of lenalidomide is 25 mg/day. It is taken orally on days D1 until D14 and on days D22 until D35 in the cycles 1 to 4. In the cycles 5 to 18 and subsequent it is taken on days D1 until D21.
The dose of dexamethasone is 20 mg/day. For patients < 75 years old it is given on days D1, D2, D4, D5, D8, D9, D11, D12, D15, D22, D23, D25, D26, D29, D30, D32 and D33 in the cycles 1 to 4. For patients ≥ 75 years old it is given on days D1, D4, D8, D11, D15, D22, D25, D29, and D32 in the cycles 1 to 4. In the cycles 5 to 17 and subsequent it is given once weekly on days D1, D8, D15, and D22 of each cycle for both age groups. In all cycles dexamethasone is either administered intravenously on the days of SARCLISA infusions or taken orally on the other days.
Each treatment cycle consists of a 28-day period. Treatment is repeated until disease progression or unacceptable toxicity.
For other medicinal products that are administered with SARCLISA, see section 5.1 and the respective current package insert – please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information.
The administration schedule must be carefully followed. If a planned dose of SARCLISA is missed, administer the dose as soon as possible and adjust the treatment schedule accordingly, maintaining the treatment interval.
Dose adjustments
No dose reduction of SARCLISA is recommended.
Administration adjustments should be made if patients experience infusion reactions (see “Method of administration” below).
For other medicinal products that are administered with SARCLISA, the respective package insert should be considered.
Special populations
Elderly
Based on population pharmacokinetic analysis, no dose adjustment is recommended in elderly patients.
Patients with renal impairment
Based on population pharmacokinetic analysis and on clinical data, no dose adjustment is recommended in patients with mild to severe renal impairment including end-stage renal disease (see section 5.2 – please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information).
Patients with hepatic impairment
Based on population pharmacokinetic analysis, no dose adjustment is recommended in patients with mild hepatic impairment. Data in patients with moderate and severe hepatic impairment are limited (see section 5.2 – please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information), but there is no evidence to suggest that dose adjustment is required in these patients.
Paediatric population
The safety and efficacy of SARCLISA in children below 18 years of age have not been established. Outside its authorized indications, SARCLISA has been studied in children aged 28 days to less than 18 years of age with relapsed or refractory acute lymphoblastic or myeloid leukaemia but efficacy has not been established. Currently available data are described in sections 4.8, 5.1 and 5.2 – please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information.
Method of administration
SARCLISA is for intravenous use. For instructions on dilution of the medicinal product before administration, see section 6.6 – please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information.
Infusion rates
Following dilution, the SARCLISA infusion should be administered intravenously at the infusion rate presented in Table 2 below (see section 5.1 – please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information). Incremental escalation of the infusion rate should be considered only in the absence of infusion reactions (see section 4.8 – please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information).
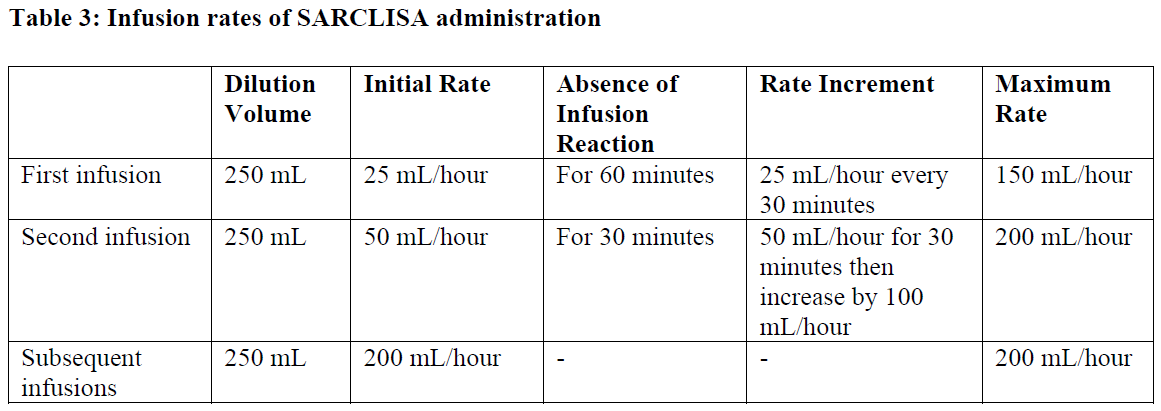
Administration adjustments should be made if patients experience infusion reactions (see section 4.4 – please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information)
In patients necessitating an intervention (Grade 2, moderate infusion reactions), a temporary interruption in the infusion should be considered and additional symptomatic medicinal products can be administered. After symptom improvement to grade ≤1 (mild), SARCLISA infusion may be resumed at half of the initial infusion rate under close monitoring and supportive care, as needed. If symptoms do not recur after 30 minutes, the infusion rate may be increased to the initial rate, and then increased incrementally, as shown in Table 2.
If symptoms do not resolve rapidly or do not improve to Grade ≤1 after interruption of SARCLISA infusion, persist or worsen despite appropriate medicinal products, or require hospitalization or are life-threatening, treatment with SARCLISA should be permanently discontinued and additional supportive therapy should be administered, as needed.
Contraindications
4.3 Contraindications
Hypersensitivity to the active substance or to any of its excipients listed in section 6.1 – please refer to the Product Insert/Patient Information Leaflet published on HSA for the full drug information.